


生成全原子蛋白質結構在全新蛋白質設計中是一個重要挑戰。目前的生成模型在主鏈生成方面有了顯著改善,但在原子精度方面仍然難以解決,因為不同的氨基酸身份被嵌入在三維空間中原子的連續位置中。這個問題在設計功能性蛋白質(如酶和分子結合劑)時尤其重要,因為即使是微小的原子級不準確性也可能妨礙實際應用。因此,採用一種新策略來有效解決這兩個方面,同時保持精度和計算效率,是克服這一挑戰的關鍵。
目前的模型如RFDiffusion和Chroma主要集中在主鏈配置上,提供有限的原子解析度。像RFDiffusion-AA和LigandMPNN這樣的擴展試圖捕捉原子級的複雜性,但無法全面表示全原子配置。基於重疊的方法如Protpardelle和Pallatom雖然試圖接近原子結構,但面臨高計算成本和處理離散-連續交互的挑戰。此外,這些方法在序列-結構一致性和多樣性之間的平衡上也存在困難,使其在精確蛋白質設計的實際應用中不太有用。
來自加州大學伯克利分校 (UC Berkeley) 和加州大學舊金山分校 (UCSF) 的研究人員介紹了ProteinZen,這是一個兩階段的生成框架,結合了主鏈框架的流匹配和潛在空間建模,以實現精確的全原子蛋白質生成。在初始階段,ProteinZen在SE(3)空間中構建蛋白質主鏈框架,同時通過流匹配方法生成每個殘基的潛在表示。這種基礎抽象避免了原子定位和氨基酸身份之間的直接糾纏,使生成過程更加流暢。在隨後的階段中,與MLM混合的變分自編碼器 (VAE) 將潛在表示解釋為原子級結構,預測側鏈扭轉角度以及序列身份。引入的通過損失提高了生成結構與實際原子特性的一致性,確保了更高的準確性和一致性。這一新框架通過在不犧牲多樣性和計算效率的情況下實現原子級準確性,克服了現有方法的限制。
ProteinZen使用SE(3)流匹配生成主鏈框架,並使用歐幾里得流匹配來生成潛在特徵,最小化旋轉、平移和潛在表示預測的損失。混合的VAE-MLM自編碼器將原子細節編碼為潛在變數,並將其解碼為序列和原子配置。該模型的架構包含張量場網絡 (Tensor-Field Networks, TFN) 用於編碼,並修改的IPMP層用於解碼,確保SE(3)等變性和計算效率。訓練是在AFDB512數據集上進行的,該數據集是通過結合PDB聚類單體和來自AlphaFold數據庫的代表性蛋白質(最多512個殘基)精心構建的。該模型的訓練利用真實和合成數據的混合來提高泛化能力。
ProteinZen實現了46%的序列-結構一致性 (SSC),在保持高結構和序列多樣性的同時超越了現有模型。它在準確性和新穎性之間取得了良好的平衡,生成的蛋白質結構多樣且獨特,具有競爭力的精度。性能分析表明,ProteinZen在較小的蛋白質序列上表現良好,並顯示出進一步開發長距離建模的潛力。合成樣本涵蓋了各種二級結構,對α-螺旋的傾向較弱。結構評估確認生成的大多數蛋白質與已知的折疊空間對齊,同時顯示出對新折疊的泛化能力。結果顯示,ProteinZen能夠生成高精度和多樣的全原子蛋白質結構,這標誌著與現有生成方法相比的一個重要進步。
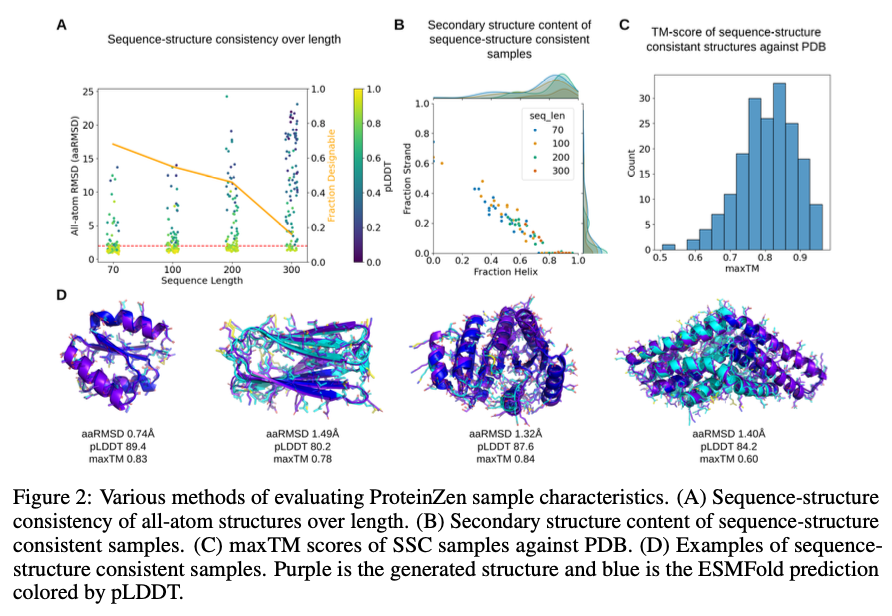
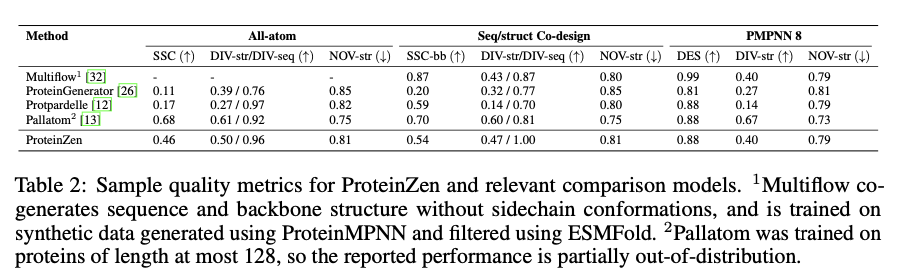
總結來說,ProteinZen通過整合SE(3)流匹配進行主鏈合成,以及潛在流匹配進行原子結構重建,介紹了一種創新的全原子蛋白質生成方法。通過將不同的氨基酸身份與原子的連續定位分開,這種技術在原子級別上達到了精確性,同時保持了多樣性和計算效率。ProteinZen的序列-結構一致性為46%,並顯示出結構獨特性,為生成蛋白質建模建立了一個新的標準。未來的工作將包括改善長距離結構建模、優化潛在空間與解碼器之間的互動,以及探索條件蛋白質設計任務。這一發展標誌著在精確、高效和實用的全原子蛋白質設計方面的重要進展。
查看論文。這項研究的所有功勞都歸於這個項目的研究人員。此外,別忘了在Twitter上關注我們,加入我們的Telegram頻道和LinkedIn小組。別忘了加入我們的60k+機器學習SubReddit。
🚨 熱門消息:LG AI研究發布EXAONE 3.5:三個開源雙語前沿AI級模型,提供無與倫比的指令跟隨和長上下文理解,為生成AI卓越的全球領導地位提供支持……。
本文由 AI 台灣 運用 AI 技術編撰,內容僅供參考,請自行核實相關資訊。
歡迎加入我們的 AI TAIWAN 台灣人工智慧中心 FB 社團,
隨時掌握最新 AI 動態與實用資訊!








{kind=link}